Karishma S Amin, Partha P Banerjee
Department of Biochemistry and Molecular and Cellular Biology, Georgetown University Medical Center, Washington DC, USA
| Date of Submission | 26-Oct-2011 |
| Date of Acceptance | 11-Nov-2011 |
| Date of Web Publication | 17-Feb-2012 |
Correspondence Address:
Partha P Banerjee
Department of Biochemistry and Molecular and Cellular Biology, Georgetown University Medical Center, Washington DC
USA
Source of Support: None, Conflict of Interest: None
DOI: 10.4103/1477-3163.93000
Abstract
Epigenetic events significantly impact the transcriptome of cells and often contribute to the onset and progression of human cancers. RASSF1A (Ras-association domain family 1 isoform A), a well-known tumor suppressor gene, is frequently silenced by epigenetic mechanisms such as promoter hypermethylation in a wide range of cancers. In the past decade a vast body of literature has emerged describing the silencing of RASSF1A expression in various cancers and demonstrating its ability to reverse the cancerous phenotype when re-expressed in cancer cells. However, the mechanisms by which RASSF1A exerts its tumor suppressive properties have not been entirely defined. RASSF1A appears to mediate three important cellular processes: microtubule stability, cell cycle progression, and the induction of apoptosis through specific molecular interactions with key factors involved in these processes. Loss of function of RASSF1A leads to accelerated cell cycle progression and resistance to apoptotic signals, resulting in increased cell proliferation. In this review, we attempt to summarize the current understanding of the biological functions of RASSF1A and provide insight that the development of targeted drugs to restore RASSF1A function holds promise for the treatment of prostate cancer.
Keywords: Epigenetic silencing, prostate cancer, RASSF1A
How to cite this article:
Amin KS, Banerjee PP. The cellular functions of RASSF1A and its inactivation in prostate cancer . J Carcinog 2012;11:3
How to cite this URL:
Amin KS, Banerjee PP. The cellular functions of RASSF1A and its inactivation in prostate cancer . J Carcinog [serial online] 2012 [cited 2021 Oct 14];11:3. Available from: https://carcinogenesis.com/text.asp?2012/11/1/3/93000
Introduction
Activation of Ras signaling pathway is a major event in the process of cancer development. Mutations within the Ras proto-oncogene commonly occur in cancer, leading to its hyperactivation, aberrant growth signaling, and unchecked cell proliferation. Extracellular cues initialize Ras activation, triggering a cascade of downstream events involving specific Ras effector proteins that in turn modulate the activities of various molecular players to elicit the desired cellular effect. [1] Although Ras signaling is typically associated with oncogenic effects such as decreased apoptosis and increased cell proliferation, it is also linked to the activation of effectors involved in tumor suppression. The RASSF1 (Ras-association domain family 1) family of proteins represents a class of Ras effectors that possess tumor suppressive properties. The RASSF1 gene comprises eight exons that undergo alternative splicing mechanisms to give rise to seven different isoforms (RASSF1A-RASSF1H). [2] It is encoded by the 3p21.3 segment of the genome, a region densely populated with tumor suppressor genes and highly susceptible to deletion and/or epigenetic silencing in various cancers. [3]
RASSF1A (Ras-association domain family isoform A), a 39 kDa protein, is characterized by the presence of a Ras association domain in its C-terminal region, allowing it to weakly interact with members of the Ras superfamily of proteins. It also possesses a SARAH (Salvador-RASSF1A-Hippo) domain in this region, essential for its interaction with members of the Hippo signaling pathway such as mammalian sterile 20-like (MST) kinases and the scaffolding protein Salvador, which also possess this domain. An ataxia telagectasia mutant (ATM) kinase phosphorylation site spans residues 125-138. A cysteine-rich domain is present in the N-terminal region of RASSF1A, found to be important in mediating some of its apoptotic effects.
Hypermethylation of the RASSF1A promoter region is probably the most frequently described epigenetic inactivation event thus far in human cancers. [2],[4] RASSF1A gene methylation has been reported in at least 37 tumor types. For example, methylation of RASSF1A is found in 80% of small cell lung cancers, [5] over 60% of breast tumors, [6],[7] in 90% of liver cancers, [8],[9],[10] in 63% of pancreatic tumors, [11] in 40% of nonileal tumors, [11] in 69% of ileal tumors, [11] in 70% of primary nasopharyngeal cancers, [8] in 91% of primary renal cell carcinomas, [12] and in 62% bladder tumors. [13]
In prostate cancer, RASSF1A gene silencing is observed in over 70% of cases, comparable to the frequency of epigenetic inactivation of other well-known tumor suppressor proteins in prostate cancer, such as the DNA damage repair protein, GSTP1 (Glutathione S transferase pi). [14],[15],[16],[17] RASSF1A expression is silenced in patient-derived tumor specimens as well as various cancer cell lines such as LNCaP, PC3, DU145, 22RV1, ND-1 mainly due to promoter hypermethylation. [14] Silencing of the RASSF1A promoter through methylation is associated with advanced grade prostatic tumors, suggesting a correlation between loss of RASSF1A expression and tumor prognosis. Moreover, large numbers of prostatic intraepithelial neoplasia (PIN; precancerous lesions) samples show RASSF1A promoter hypermethylation. [18] The high frequency of RASSF1A promoter methylation in prostate cancer and in PIN lesions suggest that RASSF1A gene silencing occurs at an early stage in prostate cancer development and has lead to the investigation of its use as a biomarker for the disease. [17]
Although there is a plethora of published literature demonstrating selective methylation of the promoter of RASSF1A gene in a large numbers of tumors, the biological functions of RASSF1A are yet to be clearly defined. Therefore, in this review we will focus on the cellular functions of RASSF1A, highlighting its role as a tumor suppressor. The role played by RASSF1A in regulating important cellular processes such as cytoskeletal dynamics, cell-cycle progression, and apoptosis is discussed in the following.
Biological Functions of Rassf1a
Modulating microtubule dynamics
Microtubules are highly dynamic polymers of tubulin and form an integral part of the cytoskeletal system. Their ability to rapidly assemble and disassemble is exploited at various stages of the cell cycle, ensuring the proper segregation of sister chromatids followed by cytokinesis. RASSF1A has been found to co-localize with microtubules via a highly charged, basic region spanning amino acids 120-288 in the primary sequence of the protein. [19] Deletion analysis studies have revealed that both the N- and C-terminals of RASSF1A are essential for its interaction with microtubules. [20] Studies suggest that microtubule-associated proteins such as MAP1 and its homologue C190RF5 could play a role in mediating the interaction of RASSF1A and the microtubule network. [21]
Depending on the stage of the cell cycle, RASSF1A has been shown to be associated with different microtubular structures, indicating that a dynamic interaction exists between this tumor suppressor protein and the cytoskeletal structure. During interphase, RASSF1A is found to be tethered to cytoplasmic microtubules. It shuttles from the centrosome in prophase to the spindle poles in metaphase and anaphase, and localizes to the midzone and midbody during telophase. [19] RASSF1A is found to have a taxol-like stabilizing effect on microtubules, making them resistant to nocodazol induced destabilization. RASSF1A-induced stabilization of microtubules decreases the motility and invasiveness of cells, thereby impeding cell migration and metastasis. Knock-down of RASSF1A is associated with loss of cell-cell adhesion and the development of a fibroblast-like morphology in HeLa cells, highlighting the significance of RASSF1A association with the microtubular network in controlling cell migration. [22]
The growth inhibitory properties of RASSF1A are dependent on its ability to bind with microtubules, as a mutant lacking the tubulin association domain was unable to induce cell-cycle arrest in 293-T cells. [23] When bound to microtubules, RASSF1A promotes G1/S and mitotic arrest. Phosphorylation of RASSF1A by Aurora-A kinase on Thr 202 and Ser 203 disrupts its association with the microtubule network, allowing mitotic progression. [24] Protein kinase C dependent phosphorylation on Ser 197 and Ser 203 also modulates the ability of RASSF1A to interact with microtubules. [25] It is possible that modifications such as phosphorylation impact the affinity of RASSF1A for microtubules or bring about a conformational change whereby it can no longer establish lasting contact with the microtubules. It remains to be determined whether any modifications in addition to phosphorylation occur on RASSF1A and how they affect its interaction with the cytoskeleton.
Controlling cell cycle progression
G1/S transition:RASSF1A over-expression results in the accumulation of cells in the G1 phase of the cell cycle. This is accompanied by a decrease in the levels of Cyclin D1, a factor essential for the progression of the cell-cycle through the Rb checkpoint. The exact mechanism by which RASSF1A brings about a decline in Cyclin D1 levels is unclear. Some studies suggest a post-translational mechanism, possibly by influencing mRNA stability or translational mechanisms, with no effect on Cyclin D1 gene expression. [26] Others have found that RASSF1A overexpression suppresses Ras-induced c-Jun-NH2-kinase (JNK) phosphorylation, resulting in decreased JNK signaling and a decline in the levels of target genes such as Cyclin D1. These data imply a possible role for RASSF1A in regulating Cyclin D1 gene expression. However, it should be noted that while a decline in JNK signaling was demonstrated experimentally in RASSF1A expressing cells, only the protein expression of Cyclin D1 was assessed, which does not extrapolate to a decline in Cyclin D1 gene induction. [27] Further studies are necessary to confirm whether RASSF1A expression has an effect on Cyclin D1 gene induction or acts post-transcriptionally, by influencing message or protein stability.
In addition to negatively affecting Cyclin D1 accumulation during G1, RASSF1A is also involved in controlling Cyclin A2 levels through its interaction with E1A regulated transcription factor, p120E4F. The fraction of cells arrested in G1 upon RASSF1A overexpression increases dramatically in the presence of p120E4F, suggesting that this factor abets RASSF1A in its growth inhibitory functions. By binding to the cyclic-AMP response element (CRE) in the promoter of Cyclin A2, p120E4F transcriptionally represses this factor that is required to progress to the next stage of the cell cycle. The ability of p120E4F to repress the Cyclin A2 gene promoter is incumbent on the expression of RASSF1A, which modulates its binding to the CRE site. Knockdown experiments with siRNA against RASSF1A have demonstrated the inability of the p120E4F to bind this site within the Cyclin A2 gene, resulting in increased expression of Cyclin A2 and a concomitant rise in the number of cells undergoing G1/s transition. [28],[29]
These studies highlight the ability of RASSF1A to stall cells in the G1 phase of the cell cycle. While RASSF1A expression is lost in most cancers due to epigenetic silencing, detectable levels of RASSF1A are present in normal cells. Surely, these cells must have a mechanism by which they can overcome the G1 arrest brought about by RASSF1A. Degradation of factors such as p21, p27kip amongst numerous others, is important for the progression of one stage of the cell cycle to the next. Cells possess specific ubiquitinylating enzyme complexes that tag key factors for degradation at each stage of the cell cycle. RASSF1A is regulated in a similar manner. The Skp1-Cul1-F-box (SCF) complex is involved in the degradation of several key players, including RASSF1A, which modulate G1/S transition. RASSF1A phosphorylation by Cyclin D-CDK4 on Ser 203 during G1 is an important event in the activation of its subsequent degradation. The substrate recognition component of the SCF complex, Skp2, recognizes the phospho moiety on RASSF1A and brings the other members of the ubiquitin ligase complex in close association with RASSF1A, thereby allowing it to be ubiquitylated and degraded by the proteasome. This explains the low levels of RASSF1A present during the S phase of the cell cycle. [30]
Regulation of mitotic progression
The anaphase promoting complex (APC) is an important ubiquitin ligase complex required for cell-cycle progression during the G1 and M phases. In conjunction with its binding partners Cdh1 and Cdc20, APC targets key regulatory factors for degradation, facilitating the progression of one stage of the cell cycle to the next. In order to keep the growth promoting effects of APC in check, the cell expresses specific inhibitory proteins such as Emi1, Mad2, at each stage of the cell cycle that maintains control over APC activation by inhibiting its binding partners.
RASSF1A is recruited to the spindle poles during prometaphase through an interaction with RASSF1A binding protein 1 (RABP1), where it encounters Cdc20, and binds to it via D-box domains present in its N-terminal region. Its interaction with Cdc20 has an inhibitory effect on APC/Cdc20 activation. Cdc20 inhibition by RASSF1A allows Cyclins A and B to linger on in the cell, thereby slowing mitotic progression. RABP1 depletion results in hyper-activation of APC, which leads to the premature destruction of mitotic cyclins, indicating that RABP1 is an important mediator of RASSF1A interaction with Cdc20. [31],[32]
The mitotic blockade caused by RASSF1A is overcome by Aurora A-mediated phosphorylation of RASSF1A. Aurora A and RASSF1A interact at the mitotic spindles, where it phosphorylates Ser 203 on RASSF1A. Phosphorylation of RASSF1A obliterates its ability to interact with Cdc20, thereby relieving the RASSF1A-mediated inhibition of APC/Cdc20. [33] A recent report presents evidence to show that the activated APC/Cdc20 complex ubiquitylates RASSF1A, priming it for degradation. Thus, Aurora A kinase mediated phosphorylation of RASSF1A promotes mitotic progression by causing APC/Cdc20 activation and subsequent degradation of RASSF1A. [34]
RASSF1A is phosphorylated once again during mitosis; however, this time Aurora B is the kinase responsible. During the late stages of mitosis, RASSF1A is known to be localized to the midzone and midbody in a Syntaxin16-dependent fashion, in a manner reminiscent of RABP1 recruitment of RASSF1A to the spindle poles during early mitosis. Syntaxin16 is a t-SNARE membrane fusion protein important in the proper execution of cytokinesis. Cells depleted of Syntaxin16 fail to complete cytokinesis and are found to be multinucleated. [35]
RASSF1A-Syntaxin16 association is dependent on RASSF1A phosphorylation on Ser 203 by Aurora B kinase. Studies have shown that nonphosphorylatable mutants of RASSF1A are unable to localize to the midzone and midbody, and the cells fail to undergo proper cytokinesis, resulting in multinucleation. In the absence of this phosphorylation event, not only does Syntaxin16 fail to interact with RASSF1A, but itself is unable localize to the midzone and midbody, suggesting that it does so in a manner dependent on RASSF1A phosphorylation. This implies that Aurora B phosphorylation mediates RASSF1A-Syntaxin16 interaction, followed by their localization to the midzone and midbody, where Syntaxin16 ensures normal cell division. [35] Thus, Aurora A and B phosphorylation events, carefully timed to occur at different stages of mitosis, govern the fate of RASSF1A in a manner that allows normal mitotic progression.
In addition to phosphorylation events, ubiquitylation also plays a role in governing the effects of RASSF1A during mitotic progression. As discussed above, the SCF ubiquitin ligase complex is important in regulating RASSF1A levels during G1/S transition. During mitosis, the Cul4A-DDB1-RING complex is essential for RASSF1A degradation and mitotic progression. Cul4A, a scaffold protein, mediates interactions between the substrate adaptor DDB1, and the E2 enzyme, allowing them to establish close proximity to one another whereby the E2 can transfer a ubiquitin moiety to the substrate, RASSF1A, bound to DDB1. These findings delineate a mechanism by which RASSF1A is degraded during mitosis, thereby allowing its normal progression. [36]
It is interesting how the levels of RASSF1A fluctuate during the different stages of the cell cycle, perfectly coordinated to allow timely progression to the next stage. Further studies are required to elucidate how RASSF1A expression is restored after its degradation in a preceding stage of the cell cycle.
Induction of apoptosis
RASSF1A overexpression triggers apoptosis by a number of different mechanisms, predominantly involving the extrinsic cell-death pathway. While the molecular players involved in RASSF1A mediated cell death are numerous and diverse, the basic mechanism underlying their activation involves their direct or indirect interaction with one or more domains in the RASSF1A protein.
RASSF1A interactions with the mammalian sterile 20-like kinases 1 and 2 (MST1/2) have been thoroughly explored. MST1/2 belong to the family of Ser/Thr kinases and are activated by auto-phosphorylation in response to stimuli such as heat shock, serum starvation, UV irradiation etc. [37] Both MST1/2 have been shown to interact directly with RASSF1A, via SARAH domains present in the C-terminal ends of both, MST and RASSF1A.
In the absence of cellular stressors, MST1/2 are found to exist in the nonphosphorylated form, maintained by Type 2A protein phosphatases (PP2A). RASSF1A interaction leads to MST1/2 dimerization and activation by auto-phosphorylation within their activation loop on Thr 180 and Thr 183, respectively. When complexed with RASSF1A, MST1/2 dephosphorylation by PP2A is prevented, prolonging the activation of these kinases. In the absence of RASSF1A, as seen in a number of different cancers, total levels of MST1/2 are not significantly affected; however, their phosphorylation decreases dramatically, suggesting that RASSF1A is important for maintaining their phosphorylated and active form. The protection of phospho-MST from the action of phosphatases by RASSF1A is specific, as it does not prevent the dephosphorylation of other PP2A substrates like Akt and Erk. [38]
MST1 interaction with RASSF1A augments its activity, leading to increased phosphorylation of histone H2B on Ser 14, chromatin condensation, and apoptosis. In this manner, RASSF1A mediates MST1 activation and induction of apoptosis. In the absence of RASSF1A, the ability of MST1 to execute Fas-induced apoptosis is diminished, suggesting that its activation by RASSF1A is essential for its proapoptotic function. [39] Some discrepancies exist concerning the effects of RASSF1A on MST1 activation. Studies have shown that RASSF1A, along with NORE1A, act as potent inhibitors of MST1 activation, which is contrary to the findings described above. [40] Experimental conditions and methodologies used might provide an explanation for the contrasting results. Further investigations to identify additional players involved in carrying out Fas-induced apoptosis via RASSF1A/MST1 interaction have lead to the identification of NDR kinases and co-activator MOB1 as important downstream effectors of apoptosis. NDR1/2 kinases belong to the AGC kinase subfamily, found to be involved in regulating cell cycle and apoptotic pathways. [41] Fas receptor stimulation was found to induce NDR1/2 activation in a RASSF1A/MST1-dependent manner. Knockdown of NDR1/2 diminished apoptotic induction by RASSF1A, indicating the importance of these kinases in this process. MOB1, a co-activator of NDR1/2 kinases, was found to be essential for their activation. [42]
RASSF1A has a similar activating effect on MST2, inducing its dimerization and autophosphorylation on Thr 180. [43] MST2 peptide sequence analysis has revealed that the Raf1 binding site overlaps with the RASSF1A binding site in the SARAH domain of MST2. In the absence of RASSF1A, MST2 exists in an inactive form bound to Raf1. The survival kinase Akt phosphorylates MST2 on its N- and C-terminal regions, promoting its preferential binding to Raf1 instead of RASSF1A. [44] Fas-induced apoptotic signals cause Raf1 to be displaced from its complex with MST2, which then binds preferentially to RASSF1A and activates downstream apoptotic players such as p73 and PUMA via the activation of members of the Hippo pathway like LATS1/2. While it is probable that RASSF1A mediated activation of MST2 activates pro-apoptotic pathways, it is surprising that the transcriptional co-activator YAP is involved in p73 activation and PUMA up-regulation, since several studies have shown that it YAP has oncogenic properties. [45],[46] In addition to Fas, DNA damage has also been shown to trigger RASSF1A/MST2-induced apoptosis. DNA damaged is perceived by the master sensor, ATM kinase, which phosphorylates RASSF1A on Ser 131 within its ATM consensus sequence, and triggers activation of MST2 and downstream events described above. [47]
Recently it was shown that RASSF1A can induce p73 activation and PUMA up-regulation by a Hippo pathway-independent mechanism, via its interaction with scaffolding protein Salvador. An RASSF1A mutant, unable to bind to Hippo pathway members including MST2, bound to Salvador and activated p73, questioning the involvement of MST2 and YAP in RASSF1A up-regulation of PUMA. [48]
RASSF1A promotes apoptosis in an MST1/2 independent manner as well. Death receptor stimulation recruits modulator of apoptosis-1 (MOAP1), a BH3-only pro-apoptotic protein, to form a TNFR-Moap1 complex, which is followed by MOAP1 engagement of RASSF1A, leading to the formation of a trimeric complex and localization of MOAP1 and RASSF1A to the activated death receptor. [49] RASSF1A-MOAP1 interaction triggers a conformational change in MOAP1, exposing its buried BH3-like domain and allowing it to interact with pro-apoptotic protein, Bax. In this manner, RASSF1A mediates Bax activation by MOAP1, inducing a conformational change in Bax that allows it to insert itself into the mitochondrial membrane and execute cell-death promoting events. In the absence of RASSF1A, Bax activation via the death receptor pathway is inhibited, suggesting that RASSF1A is an important mediator of the downstream effects of extrinsic apoptotic pathways. [50]
As described earlier, phosphorylation of RASSF1A has been shown to have important effects in regulating its association with microtubules and determining its fate during mitosis. Interestingly, RASSF1A phosphorylation also affects its apoptotic function. Protein kinase A (PKA) mediated phosphorylation of RASSF1A on Ser 203 contributes to its apoptotic effects via mechanisms involving Bax and p21. The exact mechanism by RASSF1A phosphorylation by PKA triggers apoptosis has not been described, but is thought to involve components of the Hippo pathway. [51]
On the other hand, phosphorylation of RASSF1A could also have antiapoptotic effects. It has been shown that phosphorylation of RASSF1A on Ser 175, 178, 179 by GSK3β leads to its sequestration by the 14-3-3, a member of a family of proteins that acts as molecular sponges, associating with a wide range of proteins including several pro-apoptotic proteins. Death receptor stimulation leads to a release of RASSF1A from the molecular clutches of 14-3-3, following which it is free to associate with MOAP1 and mediate Bax activation. [52]
Concluding Remarks and Future Perspectives
Since its cloning and characterization in the year 2000 by Dammann et al., [53] the status of RASSF1A expression has been investigated in a wide variety of cancer types and a significant amount of work has been conducted to establish its functions in normal cells. It is clear that RASSF1A is involved in modulating important cellular functions, as loss of its expression has been linked to pathologies such as cancer. The intricate involvement of RASSF1A in cell-cycle progression and apoptosis is evident as cells lacking its expression are unable to retain control on either process, thereby losing the delicate balance between cell growth and death.
Due to its ability to control cytoskeletal dynamics, cell-cycle progression, mitotic events and apoptosis [Figure 1], RASSF1A re-expression appears to hold promise for the treatment of cancer. Various approaches aimed at restoring RASSF1A expression in cells have been assessed. 5-aza-2′deoxycytidine, a potent DNA methyltransferase (DNMT) inhibitor, has been shown to increase levels of RASSF1A by reversing the hypermethylated state of its promoter. Although this approach successfully restores RASSF1A expression in a number of different cancer cell lines its use in the clinic should be advocated with caution as use of a nonspecific DNMT inhibitor poses the risk of reversing essential epigenetic modifications across the genome, presenting the risk of potential genomic instability. [12],[13],[15],[54],[55] Future investigations are necessary to determine whether inhibition of any particular DNMT (DNMT1, DNMT3a or DNMT3b) will be sufficient to restore RASSF1A expression. Drugs that specifically reinstate RASSF1A expression in cancer cells hold promise for the treatment of prostate cancer. We have recently shown that mahanine, an alkaloid derivative isolated from Indian curry leaves (Murraya koenigii) restores RASSF1A expression in prostate, breast, ovarian, lung, skin, and pancreatic cancer cells by down-regulating DNMT activity. [56] Similar effects were observed in prostate cancer cells treated with a synthetic fluorescent analogue (Ked-4-69) of mahanine. [57] Mahanine and Ked-4-69 appear to judiciously inhibit DNMT translocation. [57] However, further studies are necessary to confirm whether mahanine or its analogs restore RASSF1A without disrupting epigenetic modifications across the genome.
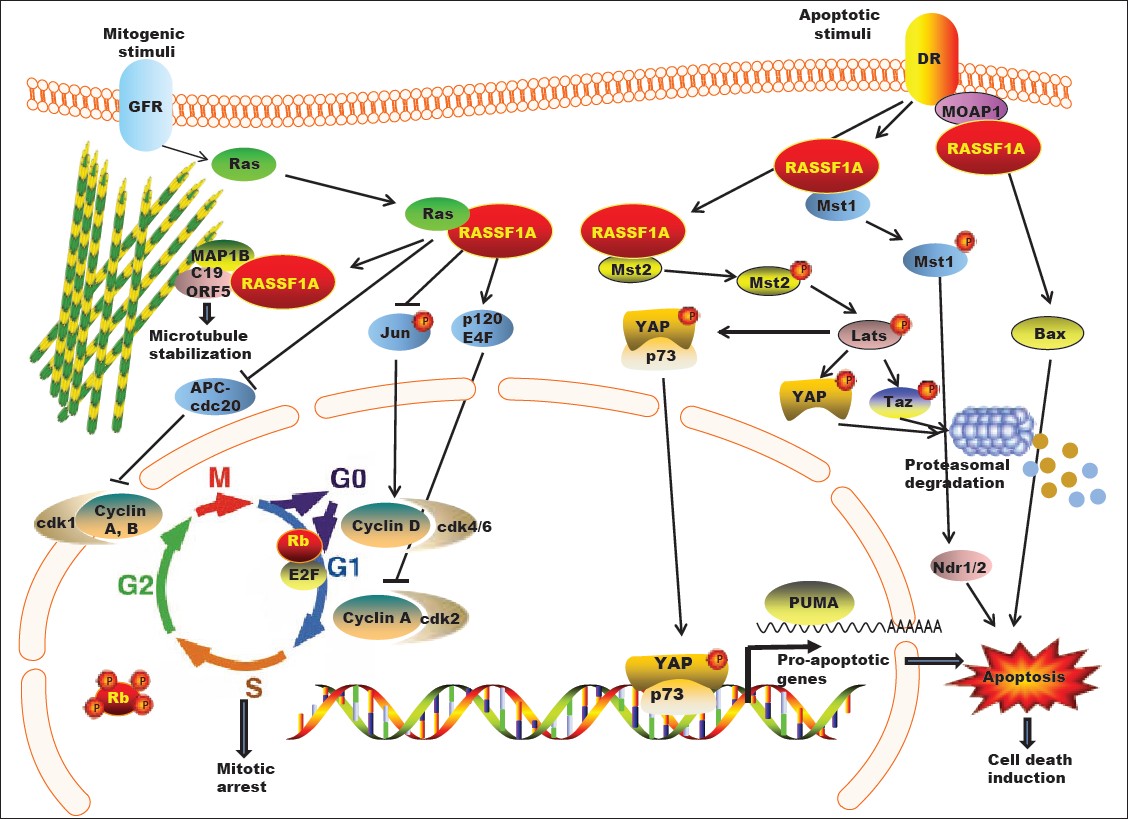 |
Figure 1: Cellular functions of RASSF1A. RASSF1A regulates microtubule dynamics, cell-cycle progression and apoptosis via its ability to engage in molecular interactions with multiple binding partners in the cell. In response to mitogenic stimuli, RASSF1A exerts inhibitory effects on G1/S transition by down-regulating Cyclins A and D. Negative regulation of APC/Cdc20 interaction by RASSF1A delays mitotic progression. Death receptor activation promotes RASSF1A interaction with effectors of apoptotic pathways, such as MST1/2, MOAP1, resulting in induction of apoptosis. GFR: Growth factor receptor; DR: Death receptor Click here to view |
The above findings suggest that RASSF1A re-expression in cancer cells has tumor suppressive effects, warranting the need for further research aimed at targeted restoration of RASSF1A expression in cells where its function is lost.
References
| 1. | Downward J Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003,3:11-22.  |
| 2. | Dammann R, Schagdarsurengin U, Strunnikova M, Rastetter M, Seidel C, Liu L, et al. Epigenetic inactivation of the Ras-association domain family 1 (RASSF1A) gene and its function in human carcinogenesis. Histol Histopathol 2003;18:665-77. [PUBMED] [FULLTEXT] |
| 3. | Hesson LB, Cooper WN, Latif F. Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene 2007;26:7283-301. [PUBMED] [FULLTEXT] |
| 4. | Pfeifer GP, Yoon JH, Liu L, Tommasi S, Wilczynski SP, Dammann R. Methylation of the RASSF1A gene in human cancers. Biol Chem 2002;383:907-14. [PUBMED] |
| 5. | Dammann R, Takahashi T, Pfeifer GP. The CpG island of the novel tumor suppressor gene RASSF1A is intensely methylated in primary small cell lung carcinomas. Oncogene 2001;20:3563-7. [PUBMED] [FULLTEXT] |
| 6. | Dammann R, Yang G, Pfeifer GP. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res 2001;61:3105-9. [PUBMED] [FULLTEXT] |
| 7. | Yan PS, Shi H, Rahmatpanah F, Hsiau TH, Hsiau AH, Leu YW, et al. Differential distribution of DNA methylation within the RASSF1A CpG island in breast cancer. Cancer Res 2003;63:6178-86. [PUBMED] [FULLTEXT] |
| 8. | Lo KW, Kwong J, Hui AB, Chan SY, To KF, Chan AS, et al. High frequency of promoter hypermethylation of RASSF1A in nasopharyngeal carcinoma. Cancer Res 2001;61:3877-81. [PUBMED] [FULLTEXT] |
| 9. | Schagdarsurengin U, Wilkens L, Steinemann D, Flemming P, Kreipe HH, Pfeifer GP, et al. Frequent epigenetic inactivation of the RASSF1A gene in hepatocellular carcinoma. Oncogene 2003;22:1866-71. [PUBMED] [FULLTEXT] |
| 10. | Zhong S, Yeo W, Tang MW, Wong N, Lai PB, Johnson PJ. Intensive hypermethylation of the CpG island of Ras association domain family 1A in hepatitis B virus-associated hepatocellular carcinomas. Clin Cancer Res 2003;9:3376-82. [PUBMED] [FULLTEXT] |
| 11. | Liu L, Broaddus RR, Yao JC, Xie S, White JA, Wu TT, et al. Epigenetic alterations in neuroendocrine tumors: methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Mod Pathol 2005;18:1632-40. [PUBMED] [FULLTEXT] |
| 12. | Dreijerink K, Braga E, Kuzmin I, Geil L, Duh FM, Angeloni D, et al. The candidate tumor suppressor gene, RASSF1A, from human chromosome 3p21.3 is involved in kidney tumorigenesis. Proc Natl Acad Sci U S A 2001;98:7504-9. [PUBMED] |
| 13. | Lee MG, Kim HY, Byun DS, Lee SJ, Lee CH, Kim JI, et al. Frequent epigenetic inactivation of RASSF1A in human bladder carcinoma. Cancer Res 2001;61:6688-92. [PUBMED] [FULLTEXT] |
| 14. | Kuzmin I, Gillespie JW, Protopopov A, Geil L, Dreijerink K, Yang Y, et al. The RASSF1A tumor suppressor gene is inactivated in prostate tumors and suppresses growth of prostate carcinoma cells. Cancer Res 2002;62:3498-502. [PUBMED] [FULLTEXT] |
| 15. | Liu L, Yoon JH, Dammann R, Pfeifer GP. Frequent hypermethylation of the RASSF1A gene in prostate cancer. Oncogene 2002;21:6835-40. [PUBMED] [FULLTEXT] |
| 16. | Maruyama R, Toyooka S, Toyooka KO, Virmani AK, Zöchbauer-Müller S, Farinas AJ, et al. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res 2002;8:514-9. |
| 17. | Ahmed H. Promoter Methylation in Prostate Cancer and its Application for the Early Detection of Prostate Cancer Using Serum and Urine Samples. Biomark Cancer 2010;2010:17-33. [PUBMED] [FULLTEXT] |
| 18. | Kang GH, Lee S, Lee HJ, Hwang KS. Aberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasia. J Pathol 2004;202:233-40. [PUBMED] [FULLTEXT] |
| 19. | Rong R, Jin W, Zhang J, Sheikh MS, Huang Y. Tumor suppressor RASSF1A is a microtubule-binding protein that stabilizes microtubules and induces G2/M arrest. Oncogene 2004;23:8216-30. [PUBMED] [FULLTEXT] |
| 20. | El-Kalla M, Onyskiw C, Baksh S. Functional importance of RASSF1A microtubule localization and polymorphisms. Oncogene 2010;29:5729-40. [PUBMED] [FULLTEXT] |
| 21. | Dallol A, Agathanggelou A, Fenton SL, Ahmed-Choudhury J, Hesson L, Vos MD, et al. RASSF1A interacts with microtubule-associated proteins and modulates microtubule dynamics. Cancer Res 2004;64:4112-6. |
| 22. | Dallol A, Agathanggelou A, Tommasi S, Pfeifer GP, Maher ER, Latif F. Involvement of the RASSF1A tumor suppressor gene in controlling cell migration. Cancer Res 2005;65:7653-9. [PUBMED] [FULLTEXT] |
| 23. | Vos MD, Martinez A, Elam C, Dallol A, Taylor BJ, Latif F, et al. A role for the RASSF1A tumor suppressor in the regulation of tubulin polymerization and genomic stability. Cancer Res 2004;64:4244-50. [PUBMED] [FULLTEXT] |
| 24. | Rong R, Jiang LY, Sheikh MS, Huang Y. Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene 2007;26:7700-8. [PUBMED] [FULLTEXT] |
| 25. | Verma SK, Ganesan TS, Parker PJ. The tumour suppressor RASSF1A is a novel substrate of PKC. FEBS Lett 2008;582:2270-6. [PUBMED] [FULLTEXT] |
| 26. | Shivakumar L, Minna J, Sakamaki T, Pestell R, White MA. The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Mol Cell Biol 2002;22:4309-18. [PUBMED] [FULLTEXT] |
| 27. | Whang YM, Kim YH, Kim JS, Yoo YD. RASSF1A suppresses the c-Jun-NH2-kinase pathway and inhibits cell cycle progression. Cancer Res 2005;65:3682-90. [PUBMED] [FULLTEXT] |
| 28. | Fenton SL, Dallol A, Agathanggelou A, Hesson L, Ahmed-Choudhury J, Baksh S, et al. Identification of the E1A-regulated transcription factor p120 E4F as an interacting partner of the RASSF1A candidate tumor suppressor gene. Cancer Res 2004;64:102-7. [PUBMED] |
| 29. | Ahmed-Choudhury J, Agathanggelou A, Fenton SL, Ricketts C, Clark GJ, Maher ER, et al. Transcriptional regulation of cyclin A2 by RASSF1A through the enhanced binding of p120E4F to the cyclin A2 promoter. Cancer Res 2005;65:2690-7. [PUBMED] [FULLTEXT] |
| 30. | Song MS, Song SJ, Kim SJ, Nakayama K, Nakayama KI, Lim DS. Skp2 regulates the antiproliferative function of the tumor suppressor RASSF1A via ubiquitin-mediated degradation at the G1-S transition. Oncogene 2008;27:3176-85. [PUBMED] [FULLTEXT] |
| 31. | Song MS, Chang JS, Song SJ, Yang TH, Lee H, Lim DS. The centrosomal protein RAS association domain family protein 1A (RASSF1A)-binding protein 1 regulates mitotic progression by recruiting RASSF1A to spindle poles. J Biol Chem 2005;280:3920-7. [PUBMED] [FULLTEXT] |
| 32. | Song MS, Song SJ, Ayad NG, Chang JS, Lee JH, Hong HK, et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat Cell Biol 2004;6:129-37. [PUBMED] [FULLTEXT] |
| 33. | Song SJ, Song MS, Kim SJ, Kim SY, Kwon SH, Kim JG, et al. Aurora A regulates prometaphase progression by inhibiting the ability of RASSF1A to suppress APC-Cdc20 activity. Cancer Res 2009;69:2314-23. [PUBMED] [FULLTEXT] |
| 34. | Chow C, Wong N, Pagano M, Lun SW, Nakayama KI, Nakayama K, et al. Regulation of APC/C(Cdc20) activity by RASSF1A-APC/C(Cdc20) circuitry. Oncogene 2011.[In Press] |
| 35. | Song SJ, Kim SJ, Song MS, Lim DS. Aurora B-mediated phosphorylation of RASSF1A maintains proper cytokinesis by recruiting Syntaxin16 to the midzone and midbody. Cancer Res 2009;69:8540-4. [PUBMED] [FULLTEXT] |
| 36. | Jiang L, Rong R, Sheikh MS, Huang Y. Cullin-4A. DNA damage-binding protein1 E3 ligase complex targets tumor suppressor RASSF1A for degradation during mitosis. J Biol Chem 2011;286:6971-8. [PUBMED] [FULLTEXT] |
| 37. | Radu M, Chernoff J. The DeMSTification of mammalian Ste20 kinases. Curr Biol 2009;19:R421-5. [PUBMED] [FULLTEXT] |
| 38. | Guo C, Zhang X, Pfeifer GP. The tumor suppressor RASSF1A prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J Biol Chem 2011;286:6253-61. [PUBMED] [FULLTEXT] |
| 39. | Oh HJ, Lee KK, Song SJ, Jin MS, Song MS, Lee JH, et al. Role of the tumor suppressor RASSF1A in Mst1-mediated apoptosis. Cancer Res 2006;66:2562-9. [PUBMED] [FULLTEXT] |
| 40. | Praskova M, Khoklatchev A, Ortiz-Vega S, Avruch J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem J 2004;381:453-62. [PUBMED] [FULLTEXT] |
| 41. | Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 2010;11:9-22. [PUBMED] [FULLTEXT] |
| 42. | Vichalkovski A, Gresko E, Cornils H, Hergovich A, Schmitz D, Hemmings BA. NDR kinase is activated by RASSF1A/MST1 in response to Fas receptor stimulation and promotes apoptosis. Curr Biol 2008;18:1889-95. [PUBMED] |
| 43. | Guo C, Tommasi S, Liu L, Yee JK, Dammann R, Pfeifer GP. RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr Biol 2007;17:700-5. |
| 44. | Romano D, Matallanas D, Weitsman G, Preisinger C, Ng T, Kolch W. Proapoptotic kinase MST2 coordinates signaling crosstalk between RASSF1A, Raf-1, and Akt. Cancer Res 2010;70:1195-203. [PUBMED] [FULLTEXT] |
| 45. | Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, Piazzolla D, et al. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell 2007;27:962-75. [PUBMED] [FULLTEXT] |
| 46. | Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A 2006;103:12405-10. [PUBMED] [FULLTEXT] |
| 47. | Hamilton G, Yee KS, Scrace S, O’Neill E. ATM regulates a RASSF1A-dependent DNA damage response. Curr Biol 2009;19:2020-5. [PUBMED] [FULLTEXT] |
| 48. | Donninger H, Allen N, Henson A, Pogue J, Williams A, Gordon L, et al. Salvador protein is a tumor suppressor effector of RASSF1A with hippo pathway-independent functions. J Biol Chem 2011;286:18483-91. [PUBMED] [FULLTEXT] |
| 49. | Foley CJ, Freedman H, Choo SL, Onyskiw C, Fu NY, Yu VC, et al. Dynamics of RASSF1A/MOAP-1 association with death receptors. Mol Cell Biol 2008;28:4520-35. [PUBMED] [FULLTEXT] |
| 50. | Baksh S, Tommasi S, Fenton S, Yu VC, Martins LM, Pfeifer GP, et al. The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to Bax conformational change and cell death. Mol Cell 2005;18:637-50. [PUBMED] [FULLTEXT] |
| 51. | Richter AM, Schagdarsurengin U, Rastetter M, Steinmann K, Dammann RH. Protein kinase A-mediated phosphorylation of the RASSF1A tumour suppressor at Serine 203 and regulation of RASSF1A function. Eur J Cancer 2010;46:2986-95. [PUBMED] [FULLTEXT] |
| 52. | Ghazaleh HA, Chow RS, Choo SL, Pham D, Olesen JD, Wong RX, et al. 14-3-3 mediated regulation of the tumor suppressor protein, RASSF1A. Apoptosis 2010;15:117-27. [PUBMED] [FULLTEXT] |
| 53. | Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 2000;25:315-9. [PUBMED] [FULLTEXT] |
| 54. | Byun DS, Lee MG, Chae KS, Ryu BG, Chi SG. Frequent epigenetic inactivation of RASSF1A by aberrant promoter hypermethylation in human gastric adenocarcinoma. Cancer Res 2001;61:7034-8. [PUBMED] [FULLTEXT] |
| 55. | Schagdarsurengin U, Gimm O, Hoang-Vu C, Dralle H, Pfeifer GP, Dammann R. Frequent epigenetic silencing of the CpG island promoter of RASSF1A in thyroid carcinoma. Cancer Res 2002;62:3698-701. [PUBMED] [FULLTEXT] |
| 56. | Jagadeesh S, Sinha S, Pal BC, Bhattacharya S, Banerjee PP. Mahanine reverses an epigenetically silenced tumor suppressor gene RASSF1A in human prostate cancer cells. Biochem Biophys Res Commun 2007;362:212-7. [PUBMED] [FULLTEXT] |
| 57. | Sheikh KD, Banerjee PP, Jagadeesh S, Grindrod SC, Zhang L, Paige M, et al. Fluorescent epigenetic small molecule induces expression of the tumor suppressor ras-association domain family 1A and inhibits human prostate xenograft. J Med Chem 2010;3:2376-82. |
Authors
Ms. Karishma S. Amin, Department of Biochemistry and Molecular and Cellular Biology at Georgetown University Medical Center
Dr. Partha P. Banerjee, Department of Biochemistry and Molecular & Cellular Biology, Georgetown University Medical Center, 3900 Reservoir Road, NW, Washington DC
Figures